-
Shifts in Excitation Energies Induced by Hydrogen Bonding: A Comparison of the Embedding and Supermolecular Time-Dependent Density Functional Theory Calculations with the Equation-of-Motion Coupled-Cluster Results
G. Fradelos, J.J. Lutz, T.A. Wesolowski, P. Piecuch and M. Wloch
in "Progress in Theoretical Chemistry and Physics" Advances in the Theory of Quantum Systems in Chemistry and Physics, ed. P. Hoggan, E. Brändas, J. Maruani, P. Piecuch and G. Delgado-Barrio, 22 (2012), p219-248


DOI:10.1007/978-94-007-2076-3_13 | unige:17800 | Abstract | Article PDF
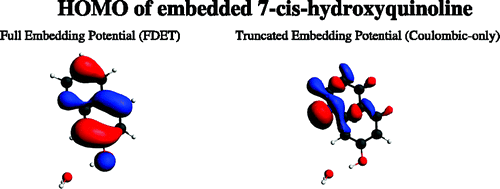
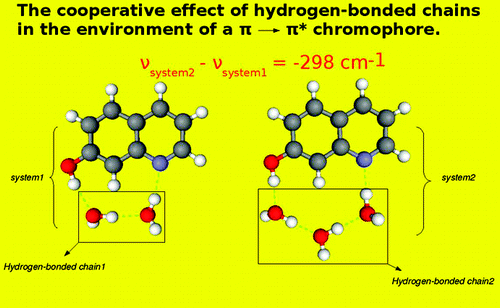
Shifts in the Ï â Ïâ excitation energy of the cis-7-hydroxyquinoline chromophore induced by hydrogen bonding with small molecules, obtained with the frozen-density embedding theory (FDET), are compared with the results of the high-level equation-of-motion coupled-cluster (EOMCC) calculations with singles, doubles, and noniterative triples, which provide the reference ab initio data, the supermolecular time-dependent density functional theory (TDDFT) calculations, and the available experimental data. It is demonstrated that the spectral shifts resulting from the FDET calculations employing nonrelaxed environment densities and their EOMCC counterparts are in excellent agreement with one another, whereas the analogous shifts obtained with the supermolecular TDDFT approach do not agree with the EOMCC reference data. Among the discussed issues are the effects of higher-order correlations on the excitation energies and complexation-induced excitation energy shifts resulting from the EOMCC calculations, and the choice of the approximants that represent the nonadditive kinetic energy contributions to the embedding potential of FDET.